NF1 is een genetische aandoening en hoort bij de neurocutane aandoeningen. Ongeveer 1 op de 2500–3000 mensen heeft NF1. De oorzaak is een fout (mutatie) in het NF1-gen.
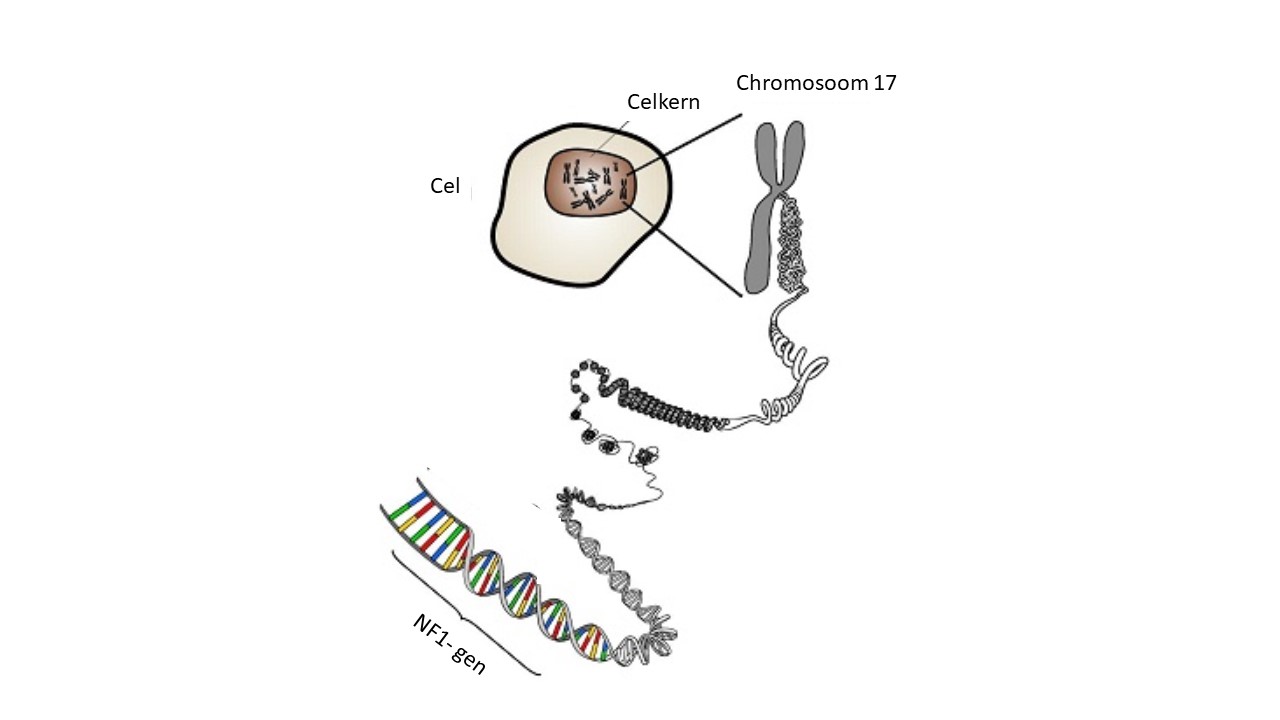
Wat doet het NF1-gen?
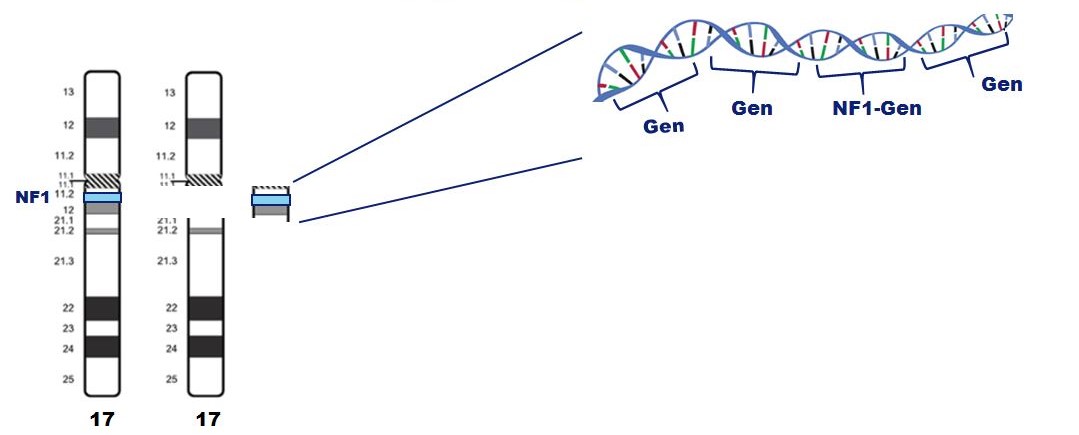
Het NF1-gen ligt op chromosoom 17 en zorgt voor het eiwit neurofibromine.
Dit eiwit remt het RAS-eiwit, dat zorgt voor celgroei.
Bij NF1 werkt neurofibromine niet goed of is er te weinig van. Daardoor remt het RAS-eiwit niet genoeg. Het RAS-eiwit wordt dan te actief, waardoor cellen onbeperkt kunnen groeien. Hierdoor ontstaan vooral goedaardige gezwellen in de huid, maar soms ook in andere delen van het lichaam.
De mutatie in het NF1-gen speelt ook een rol bij cognitieve, gedrags-, leer- en psychosociale problemen. Waarschijnlijk beïnvloeden neurofibromine en het RAS-eiwit de communicatie tussen hersencellen.
Subtypen van NF1
Subtypen van NF1
1 Gegeneraliseerde NF1
De mutatie zit in alle lichaamscellen.
2 Mozaïek NF1 (segmentale NF1)
De mutatie zit in een deel van de lichaamscellen.
Klachten beperken zich vaak tot één lichaamsdeel.
Deze vorm komt minder vaak voor en verloopt meestal milder.
3 NF1-deletie (type 1)
Bij sommige patiënten ontbreekt het hele NF1-gen (microdeletie).
Deze groep heeft vaak ernstiger klachten, zoals plexiforme neurofibromen en leerproblemen.
Ernst van de aandoening
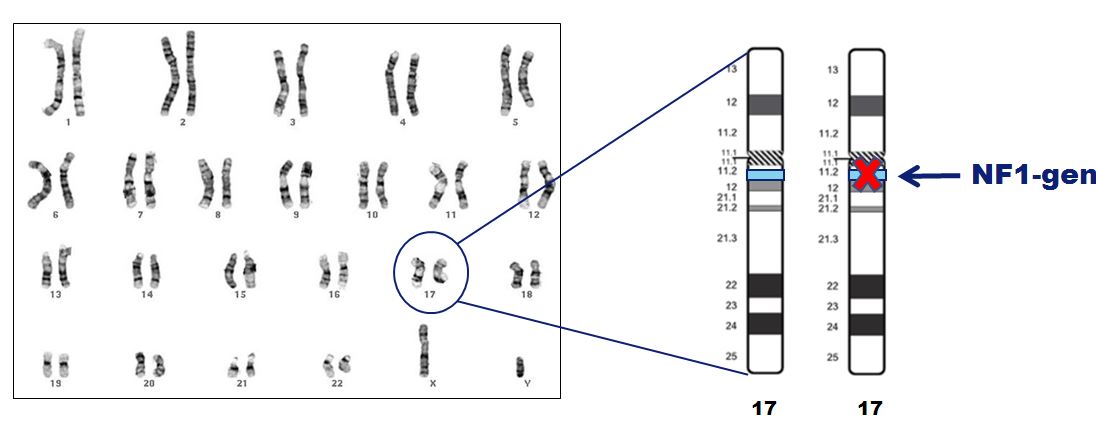
Er zijn veel verschillende mutaties in het NF1-gen. Bij slechts enkele mutaties lijkt er een verband te zijn met de ernst van NF1. Een voorbeeld: bij patiënten met een volledige deletie van het NF1-gen is de aandoening vaak ernstiger.
Type mutatie in relatie tot de ernst van de aandoening
Er zijn reeds vele mutaties aangetoond die verspreid liggen over het NF1-gen. Slechts voor enkele mutaties zijn er aanwijzingen dat er misschien een verband is tussen de mutatie en de ernst van de ziekte. Zo is bijvoorbeeld bekend dat bij NF1 patiënten bij wie het NF-1 gen volledig ontbreekt (microdeletie), de aandoening zich vaker ernstiger presenteert met bijvoorbeeld (plexiforme) neurofibromen en leerproblemen.
Erfelijkheid van NF1
NF1 wordt in de helft van de gevallen van ouder op kind doorgegeven.
In de andere helft ontstaat NF1 door een nieuwe (spontane) mutatie.
NF1 erft autosomaal dominant over.
Dit betekent: iemand met NF1 heeft bij elk kind een kans van 1 op 2 om NF1 door te geven.
Bij een kinderwens is het belangrijk dat ouders vroeg informatie krijgen.
Erfelijkheidsonderzoek
Als een toekomstige ouder NF1 heeft of denkt drager te zijn, kunnen zij terecht bij een klinisch genetisch centrum. Onderzoek kan bestaan uit:
Kans op een kind met NF1
Ouders van een kind met een spontane mutatie hebben meestal geen verhoogde kans op een volgend kind met NF1.
Er is wel een kleine kans op kiemcel-mozaïcisme. Daarbij zit de mutatie al in de eicellen of zaadcellen.
Als een ouder segmentale NF1 (mozaïcisme) heeft, is er geen verhoogd risico op een kind met NF1.
De ernst van NF1 bij ouders zegt niets over hoe het zich bij het kind uit.
Het kan milder of ernstiger zijn.
Kinderwens en zwangerschap
Een klinisch geneticus kan uitleg geven over de keuzes bij een kinderwens. Als DNA-onderzoek nodig is, moet dit op tijd worden gestart. Er kunnen ook andere opties besproken worden, zoals:
Als de moeder NF1 heeft
Het is belangrijk dat zij goed wordt voorgelicht over risico’s in de zwangerschap. Tijdens de zwangerschap kunnen neurofibromen groter of talrijker worden. Heeft de moeder plexiforme neurofibromen in ruggenmerg of bekken, dan moet de arts extra opletten. Medicijngebruik moet soms worden aangepast of gestopt.